# 不一样的富集分析可视化
clusterProfiler自带的可视化函数barplot和dotplot其实已经够看,但是想要与众不同一点的话还是得自己动手。
# 简单一点的方案
这套方案主要是将 GO 的三类 Term:BP、MF、CC 绘制到一张图上,并支持自定义颜色。

# 加载 R 包
library(clusterProfiler) | |
library(org.Hs.eg.db) | |
library(tidyverse) | |
library(ggplot2) | |
library(ggsci) |
# 定义分析函数
# GO 富集分析 | |
GOEnrichment <- function(geneDf, pAdjustMethod = "BH", pvalueCutoff = 1, | |
qvalueCutoff = 1, topNumber = 7, csvBaseName = NULL) { | |
ego <- enrichGO(gene = geneDf$SYMBOL, | |
OrgDb = org.Hs.eg.db, | |
keyType = "SYMBOL", | |
ont = "all", | |
pAdjustMethod = pAdjustMethod, | |
pvalueCutoff = pvalueCutoff, | |
qvalueCutoff = qvalueCutoff) | |
go <- as.data.frame(ego) | |
go$Term <- paste(go$ID, go$Description, sep = ': ') | |
go <- go[order(go$ONTOLOGY, go$p.adjust, method = "radix", decreasing = c(TRUE, FALSE)), ] | |
go$Term <- factor(go$Term, levels = go$Term) | |
# Top term of each group | |
top_go <- go %>% | |
group_by(ONTOLOGY) %>% | |
slice_head(n = topNumber) %>% | |
ungroup() | |
write.csv(go, paste0("GOAll.", csvBaseName, ".csv"), row.names = FALSE) | |
write.csv(top_go, paste0("GOTop", topNumber, ".", csvBaseName, ".csv"), row.names = FALSE) | |
return(list(go, top_go)) | |
} | |
# KEGG 富集分析 | |
KEGGEnrichment <- function(geneDf, pAdjustMethod = "BH", pvalueCutoff = 1, | |
qvalueCutoff = 1, topNumber = 30, csvBaseName = NULL) { | |
ekegg <- enrichKEGG(gene = geneDf$ENTREZID, | |
organism = "hsa", | |
pAdjustMethod = pAdjustMethod, | |
pvalueCutoff = pvalueCutoff, | |
qvalueCutoff = qvalueCutoff) | |
ekegg@result$Description <- paste(ekegg@result$ID, ekegg@result$Description, sep = ': ') | |
ekegg2 <- setReadable(ekegg, OrgDb = org.Hs.eg.db, keyType = "ENTREZID") | |
top_kegg <- ekegg2[1:topNumber, ] | |
write.csv(ekegg2, paste0("KEGGAll.", csvBaseName, ".csv"), row.names = F) | |
write.csv(top_kegg, paste0("KEGGTop", topNumber, ".", csvBaseName, ".csv"), row.names = F) | |
return(list(ekegg2, top_kegg)) | |
} |
# 定义颜色和绘图函数
如果你不知道选什么颜色,这里提供两种随机颜色的方法:
# 函数一
# 随机颜色 | |
randomColor <- function(n, seed = NULL) { | |
# 如果提供了种子,则设置种子以使结果可重复 | |
if (!is.null(seed)) { | |
set.seed(seed) | |
} | |
# 获取 ggsci 包中的所有函数名 | |
all_functions <- ls("package:ggsci") | |
# 获取所有 ggsci 调色板的列表 | |
all_palettes <- all_functions[grep("^pal_", all_functions)] | |
# 从所有调色板中随机选择一个 | |
random_palette <- sample(all_palettes, 1) | |
# 使用选中的调色板函数生成一个新的函数 | |
palette_function <- do.call(random_palette, list()) | |
# 使用生成的函数生成 7 种颜色 | |
palette <- do.call(palette_function, list(7)) | |
# 从生成的颜色中随机选择 n 种 | |
colors <- sample(palette, n) | |
return(colors) | |
} |
由于 ggsci 包的调色板中的颜色数量各不相同(我已知的有 7~9),函数一的 n 值设置有一定的限制(最好 < 7),需要生成更多颜色可以使用函数二:
# 函数二
randomColor <- function(n, seed = NULL) { | |
# 如果提供了种子,则设置种子以使结果可重复 | |
if (!is.null(seed)) { | |
set.seed(seed) | |
} | |
# 获取 ggsci 包中的所有函数名 | |
all_functions <- ls("package:ggsci") | |
# 获取所有 ggsci 调色板的列表 | |
all_palettes <- all_functions[grep("^pal_", all_functions)] | |
# 初始化一个空的颜色向量 | |
all_colors <- c() | |
# 遍历所有调色板,生成所有可能的颜色 | |
for (palette in all_palettes) { | |
# 使用选中的调色板函数生成一个新的函数 | |
palette_function <- do.call(palette, list()) | |
# 获取调色板中所有颜色 | |
colors <- palette_function(7) # 使用 9 个颜色,这个值可以根据需要调整 | |
# 将颜色添加到 all_colors 向量中 | |
all_colors <- c(all_colors, colors) | |
} | |
# 从生成的所有颜色中随机选择 n 种 | |
random_colors <- sample(na.omit(all_colors), n) | |
return(random_colors) | |
} |
绘图的函数:
# 绘图 | |
plotGO <- function(ego, color = "Common", seed = NULL, facetGrid = TRUE, discreteWidth = 150, | |
title = "GO Enrichment Analysis", pFileBaseName = NULL) { | |
if (color == "Common") { | |
colors <- c("#8DA1CB", "#FD8D62", "#66C3A5") | |
} else if (color == "Random") { | |
colors <- randomColor(3, seed) | |
print(paste("Use colors:", paste(colors, collapse = " "))) | |
} | |
p <- ggplot(ego, aes(Term, Count)) + | |
geom_col(aes(fill = ONTOLOGY), width = 0.8, show.legend = TRUE) + | |
scale_fill_manual(values = colors) + | |
scale_x_discrete(labels = function(x) str_wrap(x, width = discreteWidth)) + | |
scale_y_continuous(expand = expansion(mult = c(0, 0.1))) + | |
coord_flip() + | |
theme_bw() + | |
theme(panel.grid = element_blank(), panel.background = element_rect(color = 'black', fill = 'transparent'), | |
axis.text = element_text(color = "black", size = 12), legend.text = element_text(size = 13)) + | |
labs(x = 'GO Terms', title = title) | |
# 分面 | |
if (facetGrid == "TRUE") { | |
p <- p + facet_grid(ONTOLOGY~., scale = 'free_y', space = 'free_y') | |
} | |
# ggsave(paste0("EnrichmentGO.", pFileBaseName, ".pdf"), width = 12, height = 7) | |
# ggsave(paste0("EnrichmentGO.", pFileBaseName, ".png"), width = 12, height = 7) | |
return(p) | |
} | |
plotKEGG <- function(ekegg, color = "Common", seed = NULL, facetGrid = TRUE, discreteWidth = 100, | |
title = "KEGG Enrichment Analysis", pFileBaseName = NULL) { | |
if (color == "Common") { | |
colors <- c("#8DA1CB", "#FD8D62", "#66C3A5") | |
} else if (color == "Random") { | |
colors <- randomColor(2, seed) | |
print(paste("Use colors:", paste(colors, collapse = " "))) | |
} | |
ekegg$Description <- factor(ekegg$Description, levels = ekegg$Description[order(ekegg$p.adjust)]) | |
p <- ggplot(ekegg, aes(Count, Description)) + | |
geom_col(aes(fill = p.adjust), width = 0.8, show.legend = TRUE) + | |
scale_fill_gradient(low = colors[1], high = colors[2]) + | |
labs(y = "KEGG Pathways", title = title) + | |
theme_bw() + | |
theme(axis.text = element_text(color = "black", size = 12)) | |
# ggsave(paste0("EnrichmentKEGG.", pFileBaseName, ".pdf"), width = 12, height = 7) | |
# ggsave(paste0("EnrichmentKEGG.", pFileBaseName, ".png"), width = 12, height = 7) | |
return(p) | |
} |
# 函数使用
ego <- GOEnrichment(filterUnionDEGs, csvBaseName = "filter") | |
p <- plotGO(ego[[2]], color = "Random", seed = 16, pFileBaseName = "filter") | |
p <- plotGO(ego[[2]], color = "Common", pFileBaseName = "filter") | |
p | |
ggsave(paste0("EnrichmentGO.", "filter", ".pdf"), p, width = 20, height = 15) | |
ggsave(paste0("EnrichmentGO.", "filter", ".png"), p, width = 20, height = 15) | |
ekegg <- KEGGEnrichment(filterUnionDEGs, csvBaseName = "filter") | |
p2 <- plotKEGG(as.data.frame(ekegg[2]), color = "Random", seed = 312, pFileBaseName = "filter") | |
p2 | |
ggsave(paste0("EnrichmentKEGG.", "filter", ".pdf"), p2, width = 12, height = 7) | |
ggsave(paste0("EnrichmentKEGG.", "filter", ".png"), p2, width = 12, height = 7) |
# 更加花哨一点
主要用到了 circlize 和 ComplexHeatmap 这个两个包,画成环形图
学习资料:
- https://jokergoo.github.io/circlize_book/book/
- https://jokergoo.github.io/ComplexHeatmap-reference/book/index.html
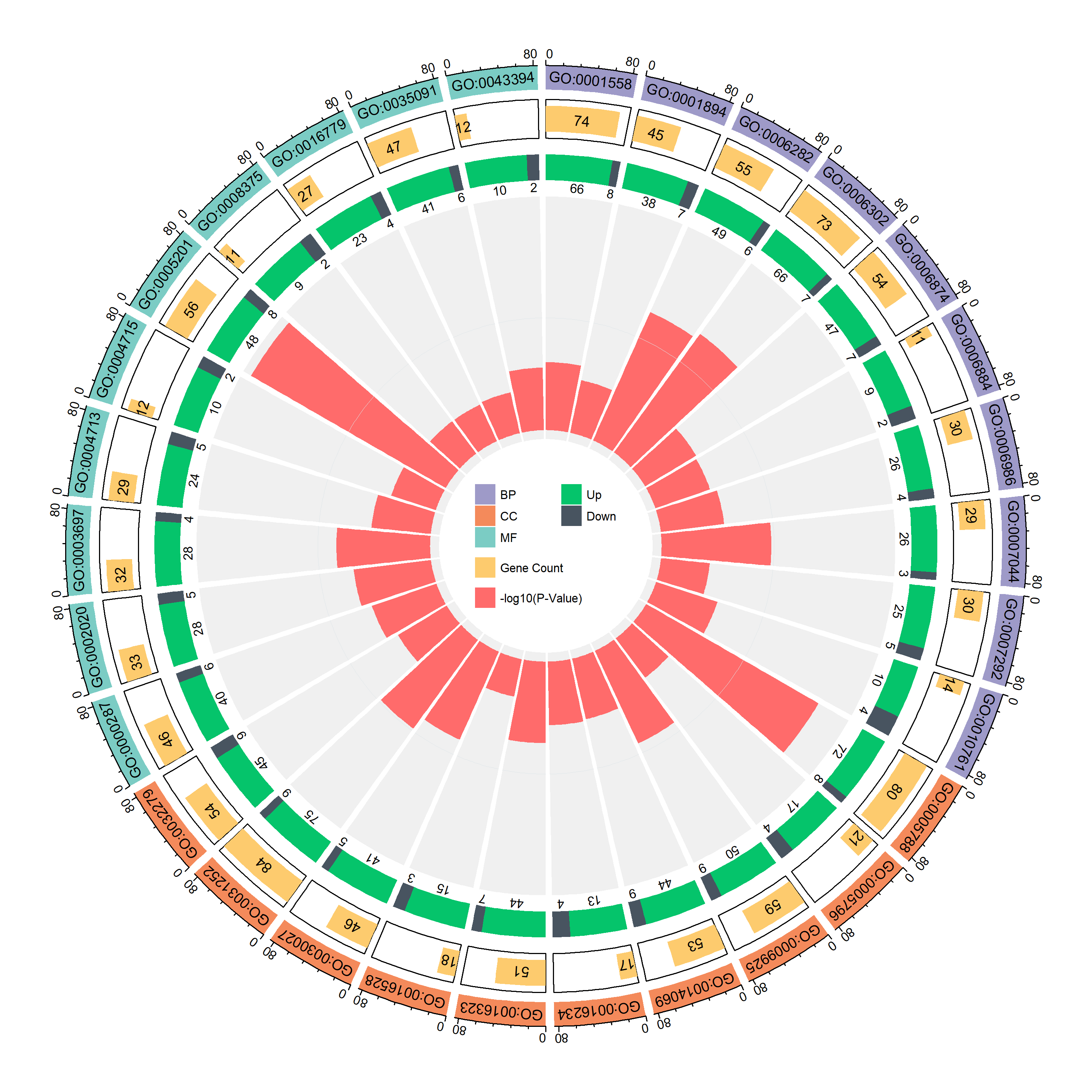
更美观的同时也展示了更多的信息:
- 最外圈是通路的类别
- 第二圈是通路基因的个数
- 第三圈是通路中上调 / 下调基因的个数
- 最内圈是 -log (P-Value) 的柱状图
# 加载 R 包
library(tidyverse) | |
library(circlize) | |
library(ComplexHeatmap) | |
library(ggsci) | |
library(Hmisc) |
# 富集分析数据准备
在这里我分别对上调、下调基因都做了富集分析(为了绘制第二圈)
# 上调 | |
intersectGene_up <- intersectGeneDf %>% | |
merge(., DEGs_TN, by.x = "SYMBOL", by.y = "Symbol") %>% | |
filter(Regulation == "Up") | |
ego_up <- GOEnrichment(intersectGene_up, topNumber = 30, csvBaseName = ".up") | |
# 下调 | |
intersectGene_down <- intersectGeneDf %>% | |
merge(., DEGs_TN, by.x = "SYMBOL", by.y = "Symbol") %>% | |
filter(Regulation == "Down") | |
ego_down <- GOEnrichment(intersectGene_down, topNumber = 30, csvBaseName = ".down") |
预处理
GO_up <- read.csv("GOAll.up.csv") | |
GO_down <- read.csv("GOAll.down.csv") | |
GO_all <- merge(GO_up, GO_down, by = "ID", suffixes = c("_up", "_down")) %>% | |
dplyr::select("ID", "ONTOLOGY_up", "Count_up", "Count_down", "pvalue_up", "pvalue_down") %>% | |
filter(pvalue_up <= 0.1, pvalue_down <= 0.1) %>% | |
group_by(ONTOLOGY_up) %>% | |
slice_head(n = 10) %>% | |
ungroup() %>% | |
mutate(total_number = Count_up + Count_down) %>% | |
mutate(Up_percent = Count_up / total_number, Down_percent = Count_down / total_number) %>% | |
mutate(start = 0, end = max(total_number)) %>% | |
mutate(total_log = (-log10(pvalue_up) + -log10(pvalue_down)) / 2) %>% | |
dplyr::select("ID", "ONTOLOGY_up", "start", "end", "total_number", "Count_up", "Count_down", "Up_percent", "Down_percent", "total_log") | |
names(GO_all)[2] <- "ONTOLOGY" | |
plot_data <- GO_all %>% | |
arrange(ONTOLOGY, ID) | |
# 将 ID 列转换为因子并按照原始顺序排序 | |
plot_data$ID <- factor(plot_data$ID, levels = plot_data$ID) |
# 圆形富集图
png("GO_enrichment_circlize.png", width = 10, height = 10, units = "in", res = 300) | |
# 颜色设置 | |
plotcolors <- list(color_BP = "#9e9ac8", color_CC = "#F48A5B", color_MF = "#7bccc4", | |
color_count = "#fdcb6e", | |
color_up = "#05c46b", color_down = "#485460", | |
color_logp = "#ff6b6b") | |
# # 可用的 seed: 1, 3, 8, 10, 12, 14, 15 down 换色 | |
# plotcolors <- as.list(randomColor(7, seed = 10)) | |
# names(plotcolors) <- c("color_BP", "color_CC", "color_MF", | |
# "color_count", | |
# "color_up", "color_down", | |
# "color_logp") | |
ONTOLOGY_Count <- table(plot_data$ONTOLOGY) %>% as.numeric() | |
GO_color <- c(rep(plotcolors$color_BP, ONTOLOGY_Count[1]), | |
rep(plotcolors$color_CC, ONTOLOGY_Count[2]), | |
rep(plotcolors$color_MF, ONTOLOGY_Count[3])) | |
circos.clear() # 确保清除之前的绘图设置 | |
circos.par("start.degree" = 90) | |
# 第一圈 GO Term | |
plot_data_1 <- plot_data %>% dplyr::select(ID, start, end) | |
circos.initializeWithIdeogram(plot_data_1, chromosome.index = plot_data_1$ID, plotType = NULL) | |
# circos.initialize(factors = plot_data_1$ID, xlim = c(0, max(plot_data_1$end))) | |
circos.track( | |
ylim = c(0, 1), | |
track.height = 0.05, # 轨道高度 | |
bg.border = NA, # 不要边框 | |
bg.col = GO_color, # 添加颜色 | |
panel.fun = function(x, y) { | |
ylim = get.cell.meta.data("ycenter") | |
xlim = get.cell.meta.data("xcenter") | |
sector.name = get.cell.meta.data("sector.index") # 提取 GO Term | |
circos.axis(h = "top", labels.cex = 0.7, major.tick.length = 0.4, labels.niceFacing = FALSE) # 刻度线 | |
circos.text(xlim, ylim, sector.name, cex = 0.8, niceFacing = FALSE) # 添加 GO Term | |
}) | |
# 第二圈 富集到的基因个数 | |
plot_data_2 <- plot_data %>% dplyr::select(ID, start, total_number) | |
# label data | |
plot_data_2_label <- plot_data_2 %>% dplyr::select(total_number) %>% as.data.frame() | |
rownames(plot_data_2_label) <- plot_data$ID | |
circos.genomicTrackPlotRegion( | |
plot_data_2, | |
track.height = 0.08, | |
bg.border = "#000000", | |
stack = TRUE, | |
panel.fun = function(region, value, ...) { | |
circos.genomicRect(region, value, col = plotcolors$color_count, border = NA, ...) | |
ylim = get.cell.meta.data("ycenter") | |
xlim = plot_data_2_label[get.cell.meta.data("sector.index"), 1] / 2 | |
sector.name = plot_data_2_label[get.cell.meta.data("sector.index"), 1] | |
circos.text(xlim, ylim, sector.name, cex = 0.8, niceFacing = FALSE) | |
}) | |
# 第三圈 上调和下调的比例 | |
plot_data_Up <- plot_data %>% | |
dplyr::select(ID, start, Up_percent, end) %>% | |
dplyr::mutate(end2 = Up_percent * end) %>% | |
dplyr::select(ID, start, end2) %>% | |
dplyr::mutate(type = 1) %>% | |
dplyr::rename(end = end2) | |
plot_data_Down <- plot_data %>% | |
dplyr::select(ID, start, Up_percent, end) %>% | |
dplyr::mutate(end2 = Up_percent * end) %>% | |
dplyr::select(ID, end2, end) %>% | |
dplyr::mutate(type = 2) %>% | |
dplyr::rename(start = end2) | |
plot_data_3 <- rbind(plot_data_Up, plot_data_Down) | |
plot_data_3_label <- plot_data %>% | |
dplyr::select(Up_percent, Down_percent, end, Count_up, Count_down) %>% | |
dplyr::mutate(Up = Up_percent * end, | |
Down = end) %>% | |
dplyr::select(Up, Down, Count_up, Count_down) %>% | |
as.data.frame() | |
rownames(plot_data_3_label) <- plot_data$ID | |
color_assign <- colorRamp2(breaks = c(1, 2), col = c(plotcolors$color_up, plotcolors$color_down)) | |
circos.genomicTrackPlotRegion( | |
plot_data_3, | |
track.height = 0.08, | |
bg.border = NA, | |
stack = TRUE, | |
panel.fun = function(region, value, ...) { | |
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...) | |
ylim = get.cell.meta.data("cell.bottom.radius") - 0.5 | |
xlim = plot_data_3_label[get.cell.meta.data("sector.index"), 1] / 2 | |
sector.name = plot_data_3_label[get.cell.meta.data("sector.index"), 3] | |
circos.text(xlim, ylim, sector.name, cex = 0.7, niceFacing = FALSE) | |
xlim = (plot_data_3_label[get.cell.meta.data("sector.index"), 2] + plot_data_3_label[get.cell.meta.data("sector.index"), 1]) / 2 | |
sector.name = plot_data_3_label[get.cell.meta.data("sector.index"), 4] | |
circos.text(xlim, ylim, sector.name, cex = 0.7, niceFacing = FALSE) | |
}) | |
# 第四圈 -log10 (pvalue) 柱状图 | |
plot_data_4 <- plot_data %>% | |
dplyr::select(ID, start, end, total_log) %>% | |
dplyr::mutate(total_log = total_log / max(total_log)) | |
circos.genomicTrack( | |
plot_data_4, | |
ylim = c(0, 1), | |
track.height = 0.5, | |
bg.col = "#f0f0f0", | |
bg.border = NA, | |
panel.fun = function(region, value, ...) { | |
sector.name = get.cell.meta.data("sector.index") | |
circos.genomicRect(region, value, col = plotcolors$color_logp, border = NA, ytop.column = 1, ybottom = 0, ...) | |
circos.lines(c(0, max(region)), c(0.5, 0.5), col = "#dfe6e9", lwd = 0.3) | |
} ) | |
circos.clear() | |
# 添加图例 | |
category_legend <- Legend( | |
labels = c("BP", "CC", "MF"), | |
type = "points", pch = NA, background = c(plotcolors$color_BP, plotcolors$color_CC, plotcolors$color_MF), | |
labels_gp = gpar(fontsize = 8), grid_height = unit(0.5, "cm"), grid_width = unit(0.5, "cm")) | |
count_legend <- Legend( | |
labels = c("Gene Count"), | |
type = "points", pch = NA, background = c(plotcolors$color_count), | |
labels_gp = gpar(fontsize = 8), grid_height = unit(0.5, "cm"), grid_width = unit(0.5, "cm")) | |
updown_legend <- Legend( | |
labels = c("Up", "Down"), | |
type = "points", pch = NA, background = c(plotcolors$color_up, plotcolors$color_down), | |
labels_gp = gpar(fontsize = 8), grid_height = unit(0.5, "cm"), grid_width = unit(0.5, "cm")) | |
logp_legend <- Legend( | |
labels = c("-log10(P-Value)"), | |
type = "points", pch = NA, background = c(plotcolors$color_logp), | |
labels_gp = gpar(fontsize = 8), grid_height = unit(0.5, "cm"), grid_width = unit(0.5, "cm")) | |
# legend_list <- lgd_list_vertical <- packLegend(category_legend, count_legend, updown_legend, logp_legend) | |
# 将图例分为三行显示 | |
lgd_list_horizontal <- packLegend( | |
category_legend, updown_legend, | |
direction = "horizontal", | |
column_gap = unit(1, "cm") | |
) | |
lgd_list_vertical <- packLegend( | |
lgd_list_horizontal, | |
count_legend, | |
logp_legend, | |
direction = "vertical", | |
row_gap = unit(0.2, "cm") | |
) | |
pushViewport(viewport(x = 0.5, y = 0.5)) | |
grid.draw(lgd_list_vertical) | |
upViewport() | |
dev.off() |
# 带基因的显著通路
对于显著的通路,我们也可以在条形图中展示少量其中的基因

# 筛选上调和下调都有的 GO Term, 每组前 10 个 | |
plot_data2 <- merge(GO_up, GO_down, by = "ID", suffixes = c("_up", "_down")) %>% | |
dplyr::select("ID", "Description_up", "Description_down", "ONTOLOGY_up", "Count_up", "Count_down", | |
"pvalue_up", "pvalue_down", "geneID_up", "geneID_down") %>% | |
filter(pvalue_up <= 0.1, pvalue_down <= 0.1) %>% | |
group_by(ONTOLOGY_up) %>% | |
slice_head(n = 10) %>% | |
ungroup() %>% | |
# mutate(total_number = Count_up + Count_down) %>% | |
# mutate(Up_percent = Count_up / total_number, Down_percent = Count_down / total_number) %>% | |
# mutate(start = 0, end = max(total_number)) %>% | |
mutate(total_log = (-log10(pvalue_up) + -log10(pvalue_down)) / 2) %>% | |
dplyr::select("ID", "Description_up", "ONTOLOGY_up", "Count_up", "Count_down", | |
"geneID_up", "geneID_down", "total_log") %>% | |
group_by(ONTOLOGY_up) %>% | |
slice_max(total_log, n = 3) %>% # 按照 total_log 列的值选择每个 ONTOLOGY_up 组中的前 3 个 | |
ungroup() | |
names(plot_data2)[2] <- "Description" | |
names(plot_data2)[3] <- "ONTOLOGY" | |
# 首字母大写 | |
plot_data2$Description <- capitalize(plot_data2$Description) | |
# 排序 | |
plot_data2$ONTOLOGY <- factor(plot_data2$ONTOLOGY) | |
# 使用排序索引重新排列数据框 | |
plot_data2 <- plot_data2[order(plot_data2$ONTOLOGY), ] | |
# terms 因子顺序 | |
plot_data2$Description <- factor(plot_data2$Description, levels = plot_data2$Description) | |
# 每个 term 展示 5 个基因 | |
plot_data2$geneID_up <- sapply(strsplit(plot_data2$geneID_up, "/"), function(x) paste(x[1:min(5, length(x))], collapse = "/")) | |
plot_data2$geneID_down <- sapply(strsplit(plot_data2$geneID_down, "/"), function(x) paste(x[1:min(5, length(x))], collapse = "/")) | |
ggplot(plot_data2, aes(x = total_log, y = rev(Description), fill = ONTOLOGY)) + | |
geom_bar(stat = "identity", width = 0.5) + | |
geom_text(aes(x = 0.1, y = rev(Description), label = Description), size = 3.5, hjust = 0) + | |
theme_classic() + | |
theme(axis.text.y = element_blank(), | |
axis.ticks.y = element_blank(), | |
axis.title.y = element_text(colour = 'black', size = 12), | |
axis.line = element_line(colour = 'black', linewidth = 0.5), | |
axis.text.x = element_text(colour = 'black', size = 10), | |
axis.ticks.x = element_line(colour = 'black'), | |
axis.title.x = element_text(colour = 'black', size = 12), | |
legend.position = "none") + | |
scale_x_continuous(expand = c(0, 0)) + | |
scale_fill_manual(values = c(plotcolors$color_BP, plotcolors$color_CC, plotcolors$color_MF)) + | |
geom_text(data = plot_data2, | |
aes(x = 0.1, y = rev(Description), label = geneID_up, color = ONTOLOGY), | |
size = 3.2, | |
fontface = 'italic', | |
hjust = 0, | |
vjust = 2.3) + | |
geom_text(data = plot_data2, | |
aes(x = 3, y = rev(Description), label = geneID_down), | |
color = "#ABB1A5", | |
size = 3.2, | |
fontface = 'italic', | |
hjust = 0, | |
vjust = 2.3) + | |
scale_color_manual(values = c(plotcolors$color_BP, plotcolors$color_CC, plotcolors$color_MF)) + | |
scale_y_discrete(expand = c(0.1, 0)) + | |
labs(title = "Enrichment of genes", | |
x = "-log10(P-Value)", | |
y = c("MF CC BP")) |